- Current software
- All software
Structural Bioinformatics
 The Paircoil2 program predicts coiled-coil domains in
protein sequences by using pairwise residue correlations
obtained from a coiled-coil database. The original Paircoil
program is still available for use.
The Paircoil2 program predicts coiled-coil domains in
protein sequences by using pairwise residue correlations
obtained from a coiled-coil database. The original Paircoil
program is still available for use.
The MultiCoil program predicts the location of coiled-coil
regions in amino acid sequences and classifies the predictions
as dimeric or trimeric. An updated version, Multicoil2, will
soon be available.
 PartiFold: Ensemble prediction of transmembrane protein structures.
Using statistical mechanics principles, partiFold computes residue
contact probabilities and sample super-secondary structures from
sequence only.
PartiFold: Ensemble prediction of transmembrane protein structures.
Using statistical mechanics principles, partiFold computes residue
contact probabilities and sample super-secondary structures from
sequence only.
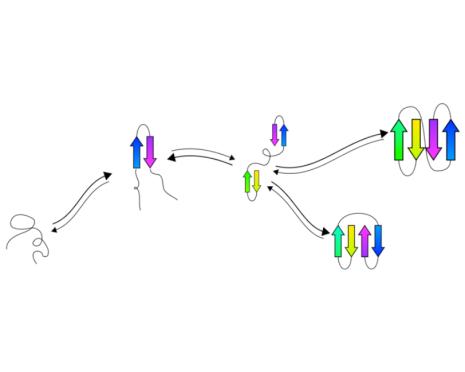 tFolder: Prediction of beta sheet folding pathways.
Predict a coarse grained representation of the folding pathway of
beta sheet proteins in a couple of minutes.
tFolder: Prediction of beta sheet folding pathways.
Predict a coarse grained representation of the folding pathway of
beta sheet proteins in a couple of minutes.
 RNAmutants: Algorithms for exploring the RNA mutational landscape.
Predict the effect of mutations on structures and reciprocally the
influence of structures on mutations. A tool for molecular evolution
studies and RNA design.
RNAmutants: Algorithms for exploring the RNA mutational landscape.
Predict the effect of mutations on structures and reciprocally the
influence of structures on mutations. A tool for molecular evolution
studies and RNA design.
 AmyloidMutants
is a statistical mechanics approach for de novo prediction and analysis of wild-type and mutant amyloid structures. Based on the premise of protein mutational landscapes, AmyloidMutants energetically quantifies the effects of sequence mutation on fibril conformation and stability.
AmyloidMutants
is a statistical mechanics approach for de novo prediction and analysis of wild-type and mutant amyloid structures. Based on the premise of protein mutational landscapes, AmyloidMutants energetically quantifies the effects of sequence mutation on fibril conformation and stability.
 MATT
is a multiple protein structure alignment program.
It uses local geometry to align segments of two sets of proteins,
allowing limited bends in the backbones between the segments.
MATT
is a multiple protein structure alignment program.
It uses local geometry to align segments of two sets of proteins,
allowing limited bends in the backbones between the segments.
 The BetaWrapPro program predicts right-handed beta-helices
and beta-trefoils by using both sequence profiles and
pairwise beta-strand interactions, and returns coordinates
for the structure.
The BetaWrapPro program predicts right-handed beta-helices
and beta-trefoils by using both sequence profiles and
pairwise beta-strand interactions, and returns coordinates
for the structure.
Genomics
RNAiCut -
Automated detection of significant genes from functional genomic screens.
 MinoTar
- Predict microRNA Targets in Coding Sequence.
MinoTar
- Predict microRNA Targets in Coding Sequence.
Systems Biology
Concordia allows you to upload an Affymetrix HGU-133 Plus 2.0 CEL file to obtain the Unified Medical Language System (UMLS) concepts that it is most enriched for based on its similarity with the microarray samples in the Concordia database.
 The Struct2Net server makes structure-based computational predictions of protein-protein interactions (PPIs), given one or two amino acid sequences as input.
The Struct2Net server makes structure-based computational predictions of protein-protein interactions (PPIs), given one or two amino acid sequences as input.
 Isobase (IsoRank PPI Network Alignment Based Ortholog Database) is a database of functionally related orthologs, which we term "isologs", developed from the multiple alignment of five major eukaryotic PPI networks, as computed by the global network alignment tools IsoRank and IsoRankN.
Isobase (IsoRank PPI Network Alignment Based Ortholog Database) is a database of functionally related orthologs, which we term "isologs", developed from the multiple alignment of five major eukaryotic PPI networks, as computed by the global network alignment tools IsoRank and IsoRankN.