Machine learning, Regulatory genomics and Disease Genetics
[Full Publication List at Google Citation ]
Roadmap Epigenomics Consortium et al. ,
Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes.
Nature: 518:317-330. (Author #8/93, equal contribution with first author in integrative analysis)
ZhiZhuo Zhang
,
Guoliang Li
,
Kim-Chuan Toh
,
Wing-Kin Sung
,
Inference of Spatial Organizations of Chromosomes Using Semi-definite
Embedding Approach and Hi-C Data.
RECOMB 2013
: 317-332
[PDF]
ZhiZhuo Zhang
,
Cheng Wei Chang
,
Hugo Willy
,
Edwin Cheung
,
Wing-Kin Sung
,
Simultaneously Learning DNA Motif along with Its Position and Sequence
Rank Preferences through EM Algorithm.
RECOMB 2012
: 355-370
[PDF]
ZhiZhuo Zhang
,
Cheng Wei Chang
,
Wan Ling Goh
,
Wing-Kin Sung
,
Edwin Cheung
,
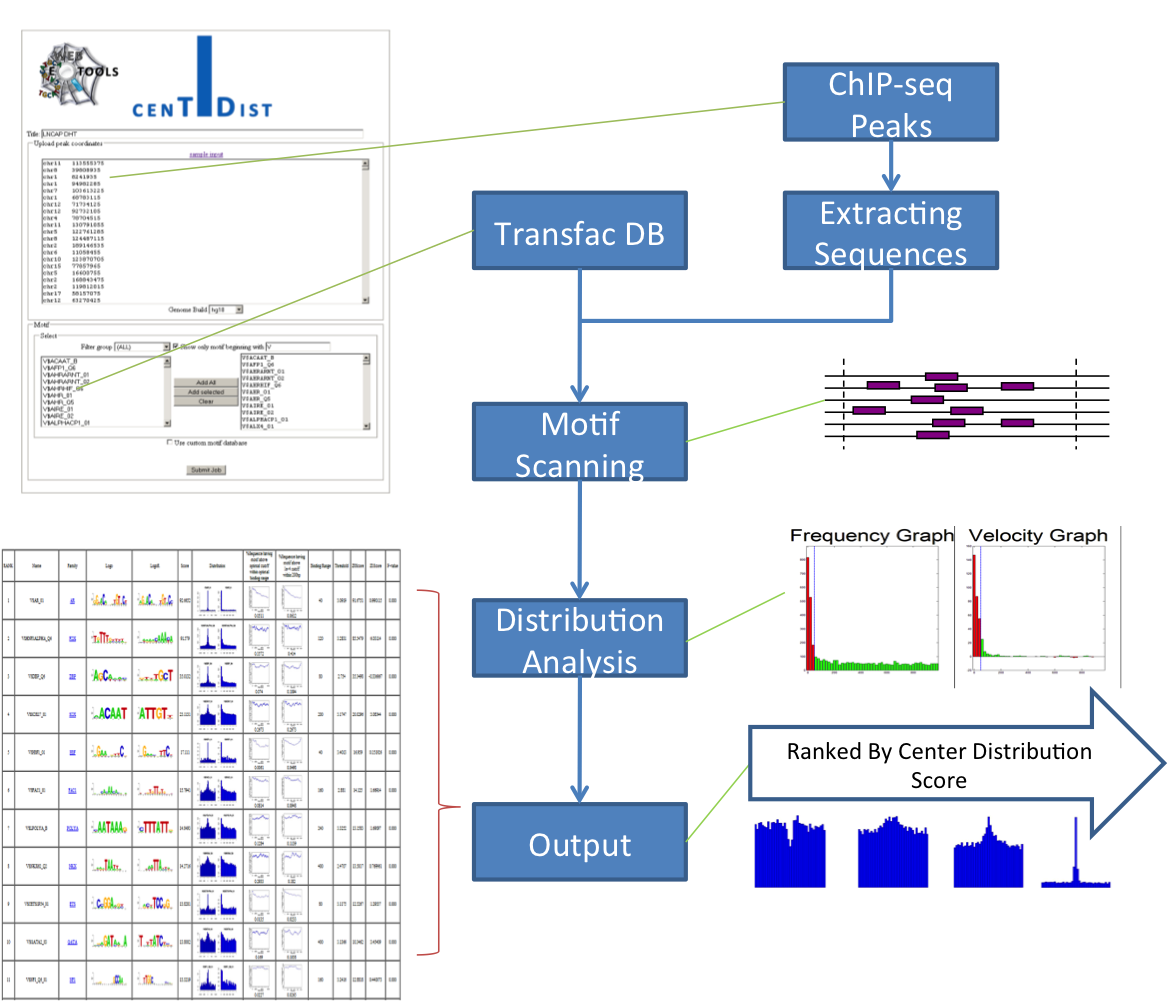
CENTDIST: discovery of co-associated factors by motif distribution.
Nucleic Acids Research 39
(Web-Server-Issue): 391-399 (2011)
[Link]
ZhiZhuo Zhang
Study of Protein-DNA Interaction Using New Generation Data.
PhD thesis, National University of Singapore.
[PDF]
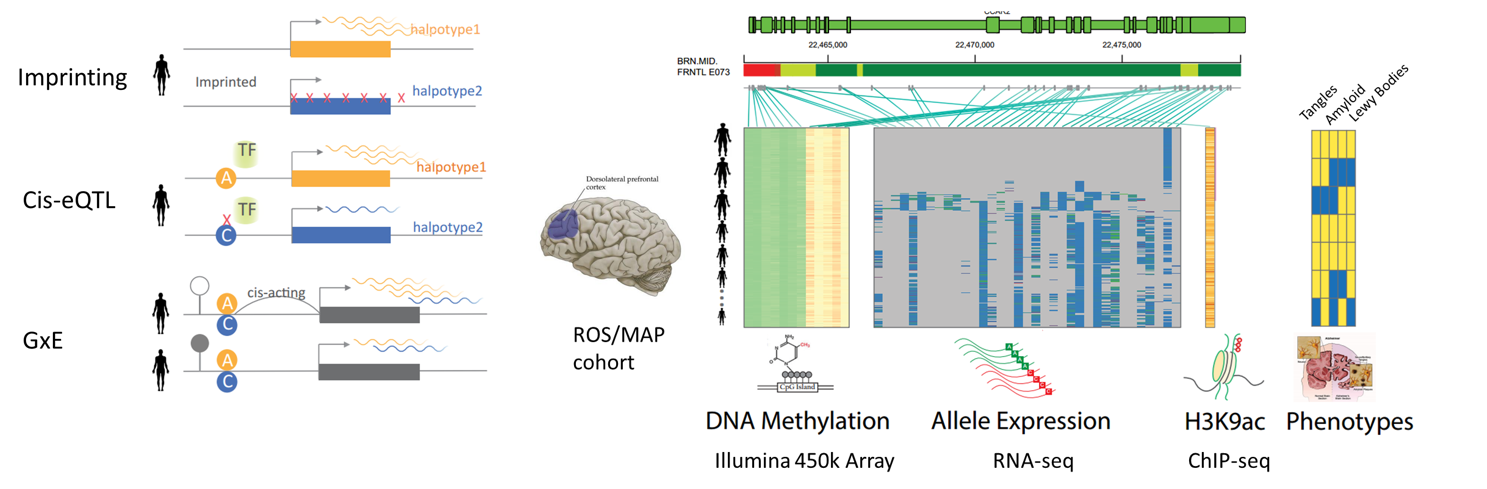
Allelic Imbalance in Aging Brain
I studied allele-specific gene expression as well as allele-specific histone modification for these set of data, and gained important insights in gene regulation and disease mechanism.
We have the most comprehensive molecular data for the largest number (~1000) of postmortem brains samples up to date, including RNA-seq, histone ChIP-seq and DNA-methylation in prefrontal cortex, as well as genotyping data.
We found evidences of imprinting genes with mono-allelic expression across individuals, including several novel imprinted genes in the aging brains. Interestingly, we showed genomic imprinting could happen in a small regulatory region instead of the whole gene body using H3K9ac ChIP-seq and DNA methylation data. Further, we noted that many susceptibility variants for CNS diseases have potential regulatory effect, which can cause allelic expression in our data. Finally, we identified novel genes whose allelic expressions are affected by brain pathologies (i.e., Amyloid Beta, Neurofibrillary Tangle,and Lewy Bodies ) through cis-variants. Our approach links cis-regulatory variants, disease pathology and target gene and pave a new way for understanding disease mechanism through allelic activity.
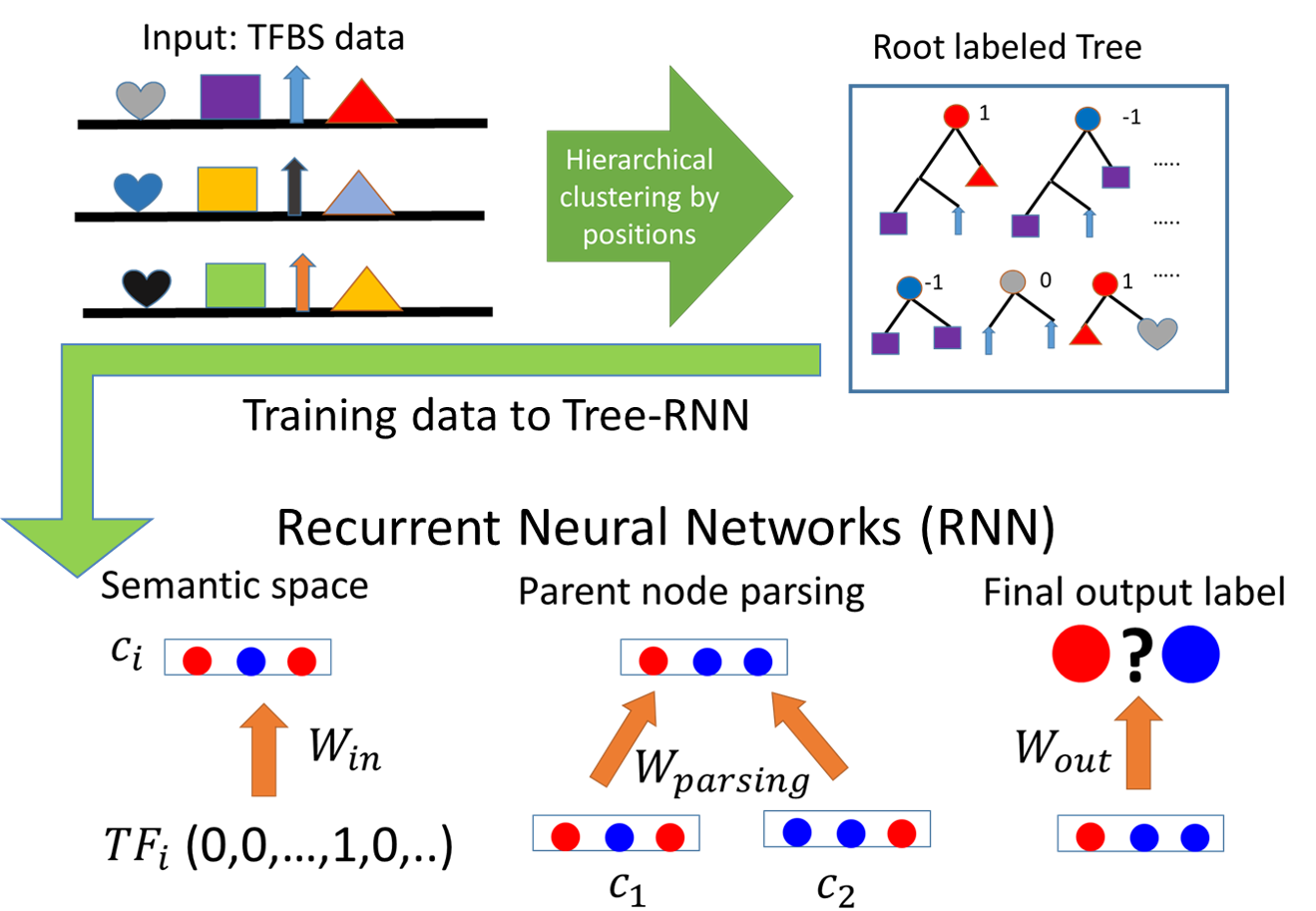
Decipher Complex Regulatory Grammar with Deep Learning
Biologically, the positions of some regulatory elements like insulators can completely change the semantic meaning of the whole regulatory region, which cannot be captured by simple grammar and shallow learning algorithms. I proposed a tree based Recurrent Neural Network (tRNN) approach, which has been successfully applied in capturing complex grammar in natural language processing. Given a list of transcription factor binding motifs (like words) on the genomic region (like a sentence), tRNN can predict the chromatin state and explore the hidden space in the neural network to identify the novel functional clusters of TF. To apply deep learning algorithm for genome-wide regulatory regions and more than 100 cell types, I developed distributed stochastic gradient descent algorithm on Apache Spark parallel computing framework and on GPUs.
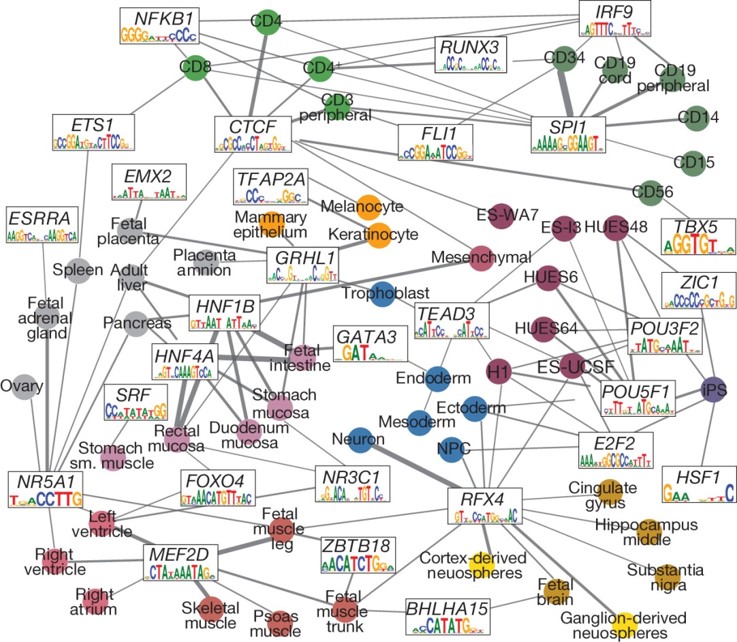
Cell Type Specific Transcription Factor Network
We sought to distinguish likely activator and repressor motifs, by identifying regulators with expression patterns across cell/tissue types that show a strong (positive or negative) correlation with the activity of enhancers in the corresponding modules9. We focused on the 40 most strongly expression-correlated regulators, and used the module-level motif enrichments to link each regulator to the cell/tissue types that define each module. We found that many of the inferred links correspond to known regulatory relationships, including OCT4 (also known as POU5F1) in pluripotent cells, HNF1B and HNF4A1 in liver and other digestive tissues, RFX4 in neurosphere and neuronal cells, and MEF2D in muscle. The most enriched regulators showed primarily positive correlations, suggesting that they function as transcriptional activators, while a subset of factors showed a negative correlation, with the motif showing enhancer depletion in the lineages where the corresponding factor is expressed, suggesting a repressive role. For example, REST (also known as NRSF), a known repressor of neuronal lineages, showed lowest expression in neuronal tissues, where its motif was most enriched in enhancers, and a similar signature was found for ZBTB1B, a known repressor of myogenesis and brain development.
Integrative Analysis of Long Range Chromatin Interactions in Prostate Cancer
An aberrant androgen receptor (AR) transcriptional network underpins prostate cancer development. While the AR cistrome has been extensively studied in prostate cancer, information pertaining to the spatial architecture of the AR transcriptional circuitry remains limited. To this end, we performed chromatin interaction analysis by paired-end tag (ChIA-PET) sequencing to profile long-range chromatin interactions associated with AR and its collaborative transcription factor, ERG, in an ERG fusion positive prostate cancer cell line. We identified ERG-associated long-range chromatin interactions as an integral component in the AR-associated chromatin interactome, acting in concert to achieve coordinated regulation of AR target genes. Through multifaceted functional data analysis, we found AR-ERG interaction hub regions are characterized by distinct functional signatures including bidirectional transcription and co-TF binding. In addition, we discovered clinically relevant lncRNAs are connected close to coding genes through AR-ERG looping. Finally, we showed prostate cancer GWAS SNPs are significantly enriched in AR-ERG co-binding sites that participate in chromatin interactions and gene regulation, suggesting identifying GWAS target genes from chromatin looping data can provide more biologically relevant findings than using the nearest gene approach. Taken together, our results revealed the presence of an AR-ERG centric higher-order chromatin structure exploited for driving prostate cancer progression.
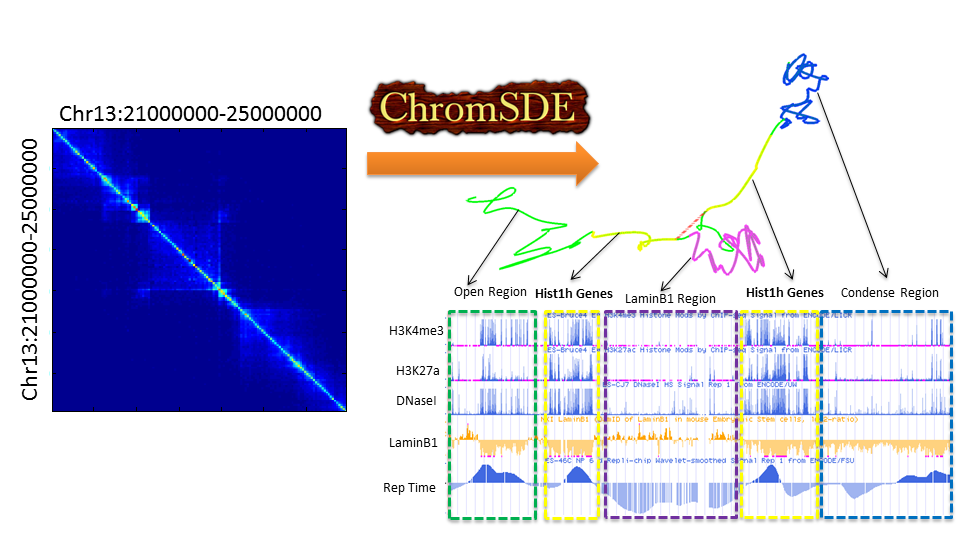
3D Chromatin Structure Modeling using Semi-definite Programming
ChromSDE is a deterministic method which applies semi-definite
programming techniques to find the best structure fitting the observed
chromatin interaction data(e.g., Hi-C data) and used golden section search to find the correct parameter for converting
the contact frequency to spatial distance. To the best of our knowledge,
ChromSDE is the only method which can guarantee recovering the correct
structure in the noise-free case. In addition, we proved that the parameter
of conversion from contact frequency to spatial distance will change under
different resolutions theoretically and empirically. Using simulation data
and real Hi-C data, we showed that ChromSDE is much more accurate and robust
than existing methods. Finally, we demonstrated that interesting biological
findings can be uncovered from our predicted 3D structure.
De novo Motif finding with positional bias and peak rank bias
Motif finding in different types of experiment data may reflect different binding preferences. We proposed a novel motif finding algorithm called SEME ( Sampling with Expectation maximization for Motif Elicitation), using full Bayesian modeling. SEME assumes the set of input sequences is a mixture of two models: a motif model and a background model. It uses unsupervised mixture model learning to learn the motif pattern (PWM), position preference and sequence rank preference at the same time; instead of asking users to provide them as inputs. SEME does not assume the presence of both preferences but automatically detect them during the motif refinement process by statistical significance testing.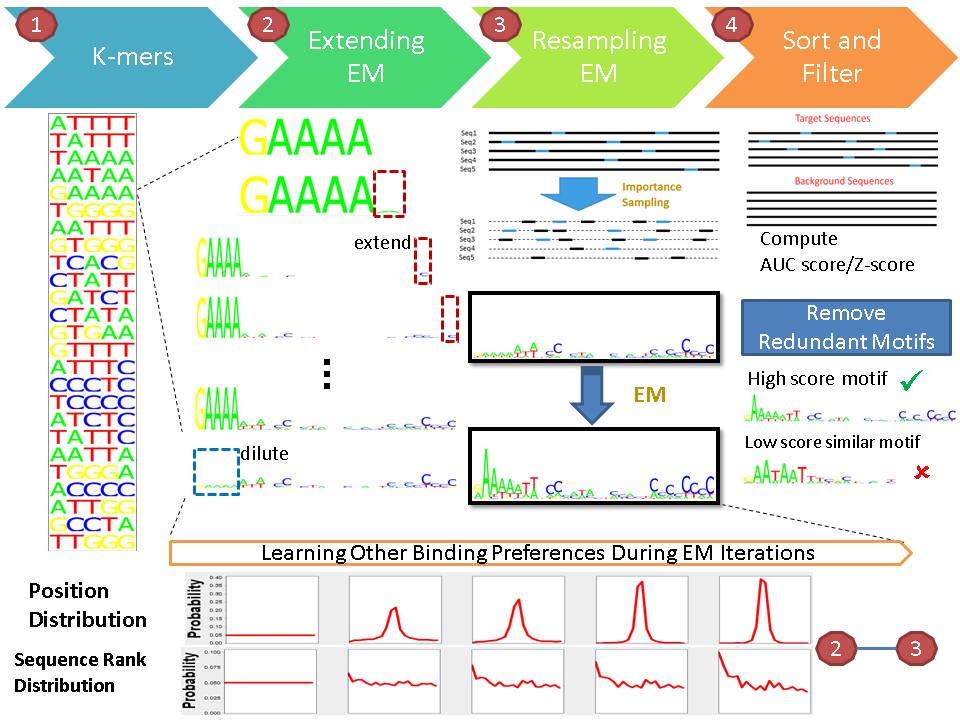
Motif enrichment of ChIP-seq Data
The main application of CENTDIST is to discover the co-TF using the known motif database, assuming that the co-TF motifs will enrich around the binding sites of ChIPed TF. Recently, several studies showed that if two TFs are co-associated, their ChIP-seq peaks (or their binding sites) are not only in close proximity with each other, but the relative distance of each TF with respect to the other exhibits a peak-like distribution. Herein, we examine whether peak-like distribution can be utilized for co-TF discovery.